
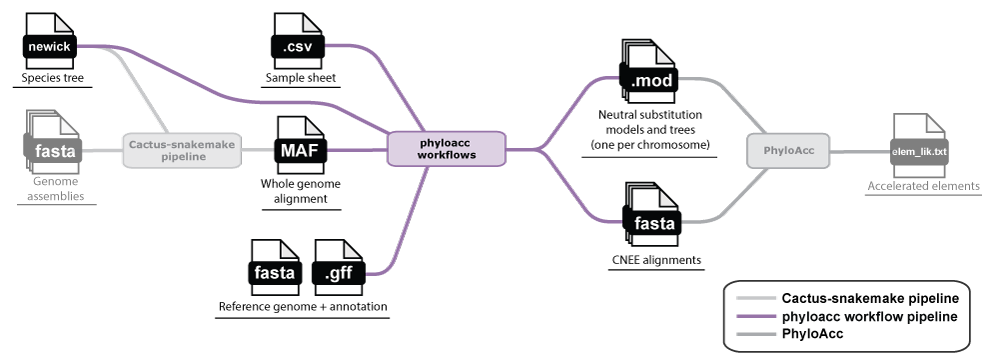
A typical set of loci for analysis with PhyloAcc are conserved non-exonic elements (CNEEs). The
phyloacc-workflows repository contains
Snakemake workflows that take a whole-genome alignment (in MAF format) and a reference genome annotation (in GFF format) and produce
a set of neutral substitution models and trees (one per scaffold/chromosome), conserved elements, and a final set of CNEE alignments ready to hand to
PhyloAcc (see the README for how PhyloAcc uses these
as input).
The pipeline works in three broad stages, each of which can be turned on or off independently in the config file:
-
Neutral model estimation: 4-fold degenerate codons are extracted from the alignment and used to fit a
neutral substitution model with
phyloFit. -
Conservation scoring: the alignment is split into manageable chunks and scored with
phastConsagainst the neutral model to call conserved regions. - CNEE extraction: conserved regions overlapping coding sequence (from a GFF) are removed, short remaining fragments are dropped, and the surviving elements are extracted as individual FASTA or MAF alignments.
The pipeline splits the alignment by chromosome/scaffold, both for scalability and to allow for chromosome-specific neutral models. This also means you will end up running PhyloAcc for each chromosome/scaffold separately, rather than on the whole genome at once.
Outputs of the pipeline include the neutral models and trees (.mod files), and the final CNEE alignments
(FASTA files).
1. Clone the repository
Use the following command to clone the repository:
git clone https://github.com/phyloacc/phyloacc-workflows.gitIf you do not have or wish to use git, download the archive directly from GitHub or with the following command:
wget https://github.com/phyloacc/phyloacc-workflows/archive/main.zip
Everything below assumes your working directory is the phyloacc-workflows directory. If you work from a different directory, you may always provide the full path to the workflow files.
2. Ensure conda is installed
The workflow uses conda to manage dependencies. You can check if conda is installed with
conda --version. If you don't have conda (i.e. if conda command
returns a "command not found" error), check out our tutorial to install it:
3. Set up the conda environment
The repository includes a small wrapper script, phyloacc_workflows, that manages a
dedicated conda environment for you with all the required dependencies. To create it, run:
./phyloacc_workflows setup
This creates a conda environment named
phyloacc-workflows.
You can confirm the environment is ready at any time with:
./phyloacc_workflows check
If you ever change envs/environment.yml or pull an update that changes it, re-running
./phyloacc_workflows setup will update the existing environment rather than recreate it
from scratch.
If ./phyloacc_workflows: Permission denied shows up when you try to run it, make the
script executable and try again:
chmod +x phyloacc_workflows
envs/environment.yml installs
snakemake-executor-plugin-slurm, so -e slurm works
out of the box. If your cluster uses a different scheduler, you'll need to
install the matching Snakemake executor plugin yourself and pass its name to -e
instead. See the
Snakemake plugin catalog
for the full list of available executors.
1. Required inputs
This workflow works in two steps: 1) predict neutral models (one per chromosome) and then 2) using the neutral models, predict conserved elements. These steps require the following inputs:
| Input | File format | Config key | Description |
|---|---|---|---|
| Whole-genome alignment | MAF | maf |
The alignment the pipeline scans for conserved elements. If you don't have one yet, see generating a whole-genome alignment in the walkthrough overview. |
| Reference genome annotation | GFF | ref_gff |
During whole genome alignment, a reference species is specified for the coordinate system in the MAF file. The GFF annotation for this species is necessary for neutral model estimation to extract 4-fold degenerate sites and to exclude coding sequence from the final CNEEs. |
| Species tree | Newick | tree_file |
The topology is used when estimating the neutral model. If you ran the
Cactus snakemake pipeline to generate your whole-genome alignment, you should
already have this. If you have a .hal file from a previous alignment, you can extract the tree with the
HAL tools command
halStats . Otherwise, you will have to infer or obtain a tree. |
The paths to these files and other pipeline options are specified in a single YAML config file, described in the pipeline configuration section.
2. Pipeline config file
Everything the workflow needs, including the paths to the raw inputs above is specified in a single YAML config file. YAML is a format
that works by pairing keys and values as key: value pairs. The keys are provided and represent
specific settings the workflow needs, and you fill in the values.
Config template
We provide a fully commented template, config-template.yaml. Copy it from the link above or the internal path below from your local copy of the repository and start filling in the paths and chromosome groups for your project:
cp config-template.yaml my-config.yaml
Open my-config.yaml in an editor. The required inputs from above are all near the top of
the file, under a section marked YOU MUST FILL THESE IN, and map onto the following config
keys, along with a few other required settings:
| Config key | Description |
|---|---|
maf |
Path to the whole-genome alignment described above. |
tree_file |
Path to the species tree described above. |
maf_ref_id |
The species label used for the reference genome in the MAF (the one whose coordinates the MAF, and ultimately the CNEEs, are reported in). See below. |
ref_gff |
Path to the GFF annotation for the reference genome described above. |
ref_chromosome_groups |
The reference chromosomes/scaffolds to analyze, organized into named groups (see below). Group names are organizational only and do not affect the analysis. They simply become subdirectories of your output. |
output_dir |
Where all workflow outputs will be written. Created automatically if it doesn't already exist. |
tmp_dir |
A directory for temporary files. Make sure it has sufficient space as whole-genome MAFs and their intermediate splits can be large. |
There are many other settings in the config file that are commented within it. In the following sections we highlight a few that are important to understand to know if you need to adjust them for your dataset.
Matching chromosome IDs
Relevant config keys: ref_chromosome_groups, maf_ref_id
maf_ref_chr_joiner, maf_chr_prefix
A common source of early errors is that the reference chromosome/scaffold IDs don't line up between the MAF and the
reference GFF. The workflow expects the IDs listed in ref_chromosome_groups to match those in
ref_gff exactly, and it derives the expected MAF src label
for each chromosome from three settings:
maf_ref_id: the reference species name as it appears in the MAF.maf_ref_chr_joiner: the character joining the reference ID and the chromosome name in the MAF (usually".").maf_chr_prefix: an optional prefix on the chromosome name in the MAF that isn't present in the GFF.
For example, if the MAF's src field looks like Homo_sapiens.chr1,
and the GFF also call that chromosome chr1, you'd set:
maf_ref_id: "Homo_sapiens"
maf_chr_prefix: ""
maf_ref_chr_joiner: "."But if the MAF instead labels it Homo_sapiens.chr1 while the GFF just calls it
1, you'd set:
maf_ref_id: "Homo_sapiens"
maf_chr_prefix: "chr"
maf_ref_chr_joiner: "."and list "1" (not "chr1") under
ref_chromosome_groups.
Splitting the alignment into chunks
Relevant config keys: split_strategy, num_seqs_max_for_gap,
num_seqs_min_gap_bp, num_seqs_min_keep_region_len
Before scoring conservation, each chromosome's alignment is split into smaller chunks, both for scalability, and
so that long stretches with little or no real alignment data don't get scored at all.
split_strategy controls how the pipeline decides where to split.
By default (split_strategy: num_seqs), the pipeline looks directly at the MAF's own
alignment blocks: any stretch where too few species are aligned is treated as a gap and used as a split point.
Three settings control split behavior:
-
num_seqs_max_for_gap(default3): a MAF block with this many species or fewer counts toward a "gap" run. -
num_seqs_min_gap_bp(default100): the minimum contiguous length of such a low-coverage run to actually count as a split point. Short blocks with low coverage may not be true split points. -
num_seqs_min_keep_region_len(default6): chunks shorter than this, after splitting, are dropped.
The num_seqs_* defaults are a starting point from exploratory analysis on one
real alignment, not a tuned recommendation. Review the number and size of chunks they produce on your own
data before trusting them.
Other split strategies include ns (split by Ns in a provided reference genome)
and fixed_windows (split into fixed-size windows). These are documented in the config template.
GC content correction of neutral models
Relevant config keys: use_gc_corrected_models, sample_file
Because the neutral models are estimated from 4-fold degenerate sites and subsequently applied to the whole genome, if those sites have different GC content the models may be inaccurate. By default, the models are corrected for this by computing GC content directly from the MAF.
Alternatively, a sample_file can be provided for GC correction.
If a column called accessions exists in the sample file, the pipeline uses
ncbi-datasets-cli to look up the GC content. If pre-computed GC values exist
in a gc column, those values are used instead.
Both columns can exist and different samples can use different methods to provide GC content.
With the GC content read, the pipeline uses PHAST's mod_freqs script to adjust neutral models for each chromosome.
Set use_gc_corrected_models: false to disable GC correction.
In many species, the GC content of 4-fold degenerate sites is similar to the genome-wide GC content, and the correction may not make a difference. However, in others (e.g. Drosophila), the 4-fold degenerate sites differ from the genome overall, and the correction is important. If you are unsure, we recommend either confirming the consistency of GC content across your genomes or just running the workflow with the correction.
And for these reasons, use_gc_corrected_models: true is the default setting in the config.
Estimating rho, or using a global value
Relevant config keys: rho_mode, fixed_rho, global_rho_stat
phastCons needs a single "rho" parameter describing how conserved the alignment is overall
relative to the neutral model, and the pipeline applies one such value per chromosome to every chunk it scores. By
default (rho_mode: fixed), that's simply the value you set for
fixed_rho (default 0.3), which we've found to be a reasonable
value for typical vertebrate datasets.
Alternatively, set rho_mode: estimate to instead have phastCons estimate rho
separately for each alignment chunk, then summarize those per-chunk estimates into a single chromosome-wide value using
global_rho_stat (p90 (value of the 90th percentile of chunk estimates) by default, or
median/mean of the chunk estimates). Any chunk whose own estimated rho exceeds that
chromosome-wide value is skipped for conservation calling, rather than scored with an inflated rho.
Filtering parameters
Relevant config keys: filter_threshold_4d, max_gap_pct,
cnee_ces_merge_gap_bp, cnee_min_len_bp
Several thresholds control how aggressively data is filtered at different stages of the pipeline (see also splitting the alignment into chunks for the settings that control where chunk boundaries themselves are placed):
-
4-fold degenerate sites (
filter_threshold_4d, default0.5): sites used to fit the neutral model are dropped if more than this fraction of sequences are missing at that site. -
Chunk quality (
max_gap_pct, default0.9): after splitting, a chunk is dropped entirely if more than this fraction of its non-reference alignment columns are gaps. -
Final CNEE filtering (
cnee_ces_merge_gap_bp, default5;cnee_min_len_bp, default50): conserved regions withincnee_ces_merge_gap_bpbp of each other (after coding sequence is removed) are merged into a single element, and anything shorter thancnee_min_len_bpbp afterward is dropped.
The defaults are reasonable for typical vertebrate-scale alignments, but you may want to loosen them for smaller or more divergent datasets, or tighten them for very large ones.
Specifying resources
Relevant config keys: rule_resources
At the bottom of the config is a list of per-rule cluster resources, which the workflow passes to Snakemake when submitting jobs.
The required resources depend on the number of species in the alignment and the size of the genomes.
Many rules are fast and light and will use the default resouces.
Others can be slow and memory-intensive and have their own resource settings.
Values in the template are based on a benchmark of a 15 species alignment of mammals. The config notes which rules should scale with genome size and which with sample size. If you run out of memory or time on a rule, increase the resources for that rule in your config file and re-run the workflow.
Full config reference
Every key recognized by the config file, in the order it appears in config-template.yaml:
| Config key | Default | Description |
|---|---|---|
maf |
Required | Path to the input MAF alignment. |
maf_ref_id |
Required | Reference species label as it appears in the MAF (see above). |
ref_gff |
Required | Path to the reference genome's GFF annotation. |
tree_file |
Required | Path to the Newick species tree. |
ref_chromosome_groups |
Required | Named groups of reference chromosomes/scaffolds to analyze. |
output_dir |
Required | Output directory for the workflow. |
tmp_dir |
Required | Directory for temporary files. |
maf_chr_prefix |
"" |
Prefix on MAF chromosome IDs not present in the GFF (see above). |
maf_ref_chr_joiner |
"." |
Character joining the reference ID and chromosome name in the MAF src field. |
filter_threshold_4d |
0.5 |
Maximum fraction of sequences allowed to be missing at a 4-fold degenerate site (see above). |
use_gc_corrected_models |
true |
Toggle GC correction of phyloFit models (see above). |
sample_file |
None; GC computed from the MAF if blank |
CSV sample sheet used for GC correction (see above). Only
relevant if use_gc_corrected_models: true, and optional even then — by
default the pipeline calculates GC content directly from the sequences in the MAF. If a sample sheet is
provided with a column called accession, GC content is instead looked up via
NCBI assembly accessions; a column called gc can be supplied instead with
precomputed values.
|
accession_header |
accession |
Column name in sample_file holding NCBI assembly accessions. |
split_strategy |
num_seqs |
num_seqs or ns (see above). |
num_seqs_max_for_gap |
3 |
Max species count for a MAF block to count toward a gap run (see above). |
num_seqs_min_gap_bp |
100 |
Minimum length of a low-coverage run to count as a split point (see above). |
num_seqs_min_keep_region_len |
6 |
Minimum chunk length (bp) to keep, split_strategy: num_seqs (see above). |
ref_fasta |
Required if split_strategy: ns |
Only needed if you set split_strategy: ns (see splitting the alignment into
chunks). This assembly is used to split the alignment into chunks based on runs of Ns; if it doesn't already
have Ns in it, the assembly must be hard masked first. |
ref_fasta_index |
Auto-generated if blank; only used if split_strategy: ns |
Path to the reference FASTA's .fai index. |
min_Ns_to_split_by |
100 |
Minimum run of Ns used as a split point, split_strategy: ns (see above). |
min_keep_region_len |
6 |
Minimum chunk length (bp) to keep, split_strategy: ns (see above). |
max_gap_pct |
0.9 |
Maximum non-reference gap fraction allowed before a chunk is filtered out (see above). |
rho_mode |
fixed |
fixed or estimate (see above). |
fixed_rho |
0.3 |
Fixed rho value used when rho_mode: fixed. |
global_rho_stat |
p90 |
Summary statistic (p90/median/mean)
used when rho_mode: estimate. |
cnee_output_format |
fasta |
Final CNEE alignment format: none, fasta, or
maf. |
cnee_ces_merge_gap_bp |
5 |
Gap (bp) allowed when merging adjacent conserved regions into a single CNEE (see above). |
cnee_min_len_bp |
50 |
Minimum length (bp) for a conserved region to be kept as a CNEE (see above). |
cnee_fasta_header |
species-coords-id |
Header format used for extracted CNEE FASTA sequences. |
cnee_expected_species |
""; read from tree_file if blank |
Optional comma-separated species list to validate CNEE FASTA extraction against. |
cnee_expected_species_file |
""; read from tree_file if blank |
Optional file with a newline-delimited species list, as an alternative to cnee_expected_species. |
rule_resources |
See above | Per-rule cluster resources (see above). |
maf_split_chr_dir |
"" |
Optional override for the chromosome-split MAF directory. |
phylofit_chr_dir |
"" |
Optional override for the chromosome-specific phyloFit model directory. |
target_ref_chromosomes |
[] |
Optional subset of chromosomes to restrict analysis to, overriding ref_chromosome_groups. |
debug_keep_intermediates |
false |
Keep intermediate files that would otherwise be cleaned up. |
cleanup_chunk_intermediates |
true |
Remove per-chunk intermediate files once a chromosome finishes. |
keep_cnee_sidecars |
false |
Keep extra per-CNEE sidecar files produced during extraction. |
run_phylofit |
true |
Enable/disable the neutral model (phyloFit) stage. |
run_phastcons |
true |
Enable/disable the phastCons conservation scoring stage. |
build_cnees |
true |
Enable/disable CNEE extraction from the conserved regions. |
display |
false |
Print the resolved config and exit, without running anything (debugging). |
version |
false |
Print the pipeline version and exit. |
info |
false |
Print pipeline meta information and exit. |
debug |
false |
Enable verbose debug logging. |
The workflow is executed through the same phyloacc_workflows wrapper used for setup. Its
run subcommand activates the conda environment and passes everything you give it straight
through to snakemake, defaulting to the Snakefile in the repository unless you specify
your own with -s.
1. Dry run
Always start with a dry run to make sure the config file is valid and to see what jobs Snakemake plans to run, before anything is actually submitted or executed:
./phyloacc_workflows run --configfile my-config.yaml -j 20 -e slurm --dryrun
Here, -j 20 is the maximum number of jobs Snakemake will have in flight at once, and
-e slurm tells Snakemake to submit jobs to a SLURM cluster using the resources you set per
rule under rule_resources in your config file. If you're testing on a single machine
instead of a cluster, drop -e slurm and Snakemake will run everything locally using up to
-j CPU cores.
If you run phyloacc_workflows run without -e/--executor
and without an active SLURM job allocation, the wrapper will print a warning: Snakemake will run every step
directly on whichever machine you launched it from. On a shared cluster login node, that means real compute
work running where it shouldn't. Either add -e slurm, or request an interactive
allocation first.
Here is an example rulegraph for fitting neutral models and extracting CNEEs
2. Executing the workflow
Once the dry run looks right, drop --dryrun to actually run it:
./phyloacc_workflows run --configfile my-config.yaml -j 20 -e slurm
Depending on the size of your alignment and how many chromosomes/scaffolds you're analyzing, this can take anywhere
from minutes to many hours. Snakemake will print progress as jobs are submitted and complete, and each rule also
writes its own log under <output_dir>/logs/<rule name>/ for closer
inspection.
3. Re-running and troubleshooting
Snakemake only re-runs rules whose outputs are missing or out of date, so if a run is interrupted or errors out, address the cause of the failure and then run the exact same command again and it will pick up where it left off rather than starting over.
If a particular rule keeps failing, check its log file first, both under
<output_dir>/logs/<rule name>/ and (for cluster runs) in the SLURM job's own
output. Common early culprits are a chromosome/scaffold ID that doesn't match between the MAF and GFF
(see Matching chromosome IDs), or a cluster partition/resource in
rule_resources that doesn't exist on your system.
All outputs are written under the output_dir you set in your config file, organized into
numbered subdirectories reflecting the stage of the pipeline that produced them. The ones you'll care about most are:
Path (relative to output_dir) |
Contents |
|---|---|
02-neutral-model/phylofit/ |
Per-chromosome neutral substitution models (.mod files) fit with
phyloFit, GC-corrected by default. |
04-phastcons/regions/<group>/<chromosome>.bed |
All conserved regions called by phastCons for that chromosome, before coding
sequence is removed. |
05-cnees/phastcons/bed/<group>/<chromosome>.cnees.bed4 |
The final CNEE coordinates for that chromosome (BED4: chromosome, start, end, CNEE ID), with coding sequence removed and short fragments filtered out. |
05-cnees/phastcons/fasta/<group>/<chromosome>/ |
One alignment file per CNEE (in the format set by cnee_output_format), plus a
manifest.txt listing them. This directory is what you point PhyloAcc's
-d option at (see the README). |
logs/ |
Per-rule log files, useful for troubleshooting failed or unexpected runs. |
From here, the CNEE alignment directory for a chromosome (or all of them pooled together) is ready to hand straight to
phyloacc.py along with the neutral model produced above. See the PhyloAcc
README for how to set up and run PhyloAcc itself on these inputs.